
La Sindrome di Lynch è una condizione genetica responsabile dell’aumento del rischio di numerose neoplasie con insorgenza, generalmente, giovanile. E’ definita anche tumore del colon non poliposico ereditario (HNPCC – dall’inglese Hereditary Non-Polyposis Colorectal Cancer) perché il tumore del colon si presenta in assenza di polipi, contrariamente a quanto accade spesso nei casi più frequenti di tumore del colon-retto.
Come si diagnostica la Sindrome di Lynch
Per sospettare la presenza di una Sindrome di Lynch si usano i criteri di Amsterdam, che si rifanno soprattutto alla storia familiare del paziente. Questi criteri vennero definiti nel 1991 ad Amsterdam e revisionati nel 1999 e sono:
- almeno 3 familiari affetti da tumori confermati istologicamente che appartengono allo spettro della sindrome HNPCC (tumori colorettali, endometriali, del piccolo intestino, delle vie urinarie);
- uno dei soggetti deve essere un parente di primo grado degli altri due su due generazioni;
- almeno uno dei tumori deve essere diagnosticato prima dei 50 anni.
Nelle famiglie identificate secondo questi criteri, i pazienti sviluppano principalmente tumori colon-rettali e/o endometriali con un rischio cumulativo di 70-80% ai 70 anni.
Nel 1997 vennero messi a punto i criteri di Bethesda, i quali tengono conto anche di altri fattori.
La Sindrome di Lynch rappresenta circa dal 2 al 4% dei casi di tumori del colon-retto e dal 2 al 3% di quelli dell’endometrio.
La Sindrome di Lynch: come si manifesta
Le principali tipologie di tumori associati alla Sindrome di Lynch sono il tumore del colon-retto, dell’endometrio e meno frequentemente, gastrico, mammario, ovarico, piccolo intestino, pancreas, prostata, tratto urinario (uretra, pelvi renale, vescica), renale, epatico e tratto biliare. Il rischio di insorgenza dei diversi tipi di tumori varia in relazione all’età con un rischio di neoplasie giovanile superiore per i soggetti con Lynch rispetto alla stessa neoplasia nella forma sporadica.
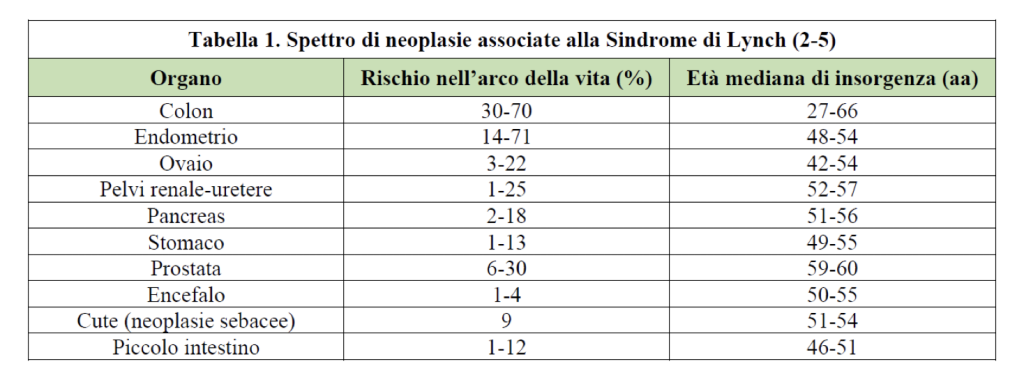
Tabella tratta da: Linee Guida Tumore del Colon – AIOM Edizione 2018
Gli aspetti genetici della Sindrome di Lynch
La Sindrome di Lynch si trasmette con modalità autosomica dominante con rischio di trasmissione alla prole del 50%. I principali geni implicati appartengono alla famiglia dei geni responsabili della riparazione dei difetti di appaiamento del DNA (DNA mismatch repair o MMR), cioè nel controllo dell’esattezza della replicazione e sono i geni MLH1, MSH2, MSH6 e PMS2. Vi è poi un quinto gene, il gene EPCAM, che non fa parte del MMR ma ne è strettamente collegato (in particolare influendo su MSH2). Vi è inoltre evidenza che il rischio di tumore del colon sembri variare in relazione al gene mutato presentando un rischio maggiore se coinvolti sono i geni MLH1, MSH2 rispetto a quando la mutazione coinvolge i geni MSH6 e PMS2.
Fondamentale risulta pertanto il percorso di consulenza oncogenetica a cui dovrebbero essere inviati i pazienti in cui è stata posta diagnosi di una delle neoplasie ascrivibili alla Sindrome di Lynch soprattutto se vi è evidenza di familiarità per le suddette neoplasie.
La Sindrome di Lynch: la gestione del follow up
Le persone con Sindrome di Lynch, sia che abbiano già manifestato una neoplasia, sia che siano asintomatiche, ovvero familiari sani in cui è stata identificata la variante genetica familiare, devono essere inseriti in un percorso specifico di follow up (sorveglianza clinico-strumentale attiva).
Le Linee Guida raccomandano che per il cancro del colon-retto, la colonscopia sia indicata a partire dai 20-25 anni di età, oppure (a seconda di quale si verifica prima) a un’età da 2 a 5 anni inferiore rispetto a quella della persona con la diagnosi più giovane in famiglia. La colonscopia, con eventuale rimozione dei polipi, andrebbe poi ripetuta ogni 1 o 2 anni.
Per le donne dai 30-35 anni in poi è raccomandata una valutazione ginecologica per la prevenzione ed eventuale identificazione precoce del carcinoma dell’endometrio e dell’ovaio. In alcune occasioni, anche sulla base della storia familiare, può essere discussa la possibilità di eseguire gastroscopia per i tumori dello stomaco, ecografia renale e esame citologico delle urine per i tumori delle vie urinarie.
In casi selezionati è possibile considerare la colectomia, cioè la rimozione di parte o di tutto il colon, mentre per le donne che hanno superato l’età riproduttiva è possibile ricorrere all’asportazione dell’utero, delle ovaie e delle tube, per evitare il tumore dell’endometrio o dell’ovaio.
Per ulteriori approfondimenti puoi contattare i genetisti di Bgenetica o, qualora dovessi rientrare in una delle condizioni prima illustrate, valutare la possibilità di eseguire una consulenza genetica, dal vivo o online.
Bibliografia
- J G Park , H F Vasen, K J Park, P Peltomaki, M Ponz de Leon, M A Rodriguez-Bigas, J Lubinski, N E Beck, M L Bisgaard, M Miyaki, J T Wijnen, S Baba, H T Lynch. Suspected hereditary nonpolyposis colorectal cancer: International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) criteria and results of genetic diagnosis. Dis Colon Rectum 1999 Jun;42(6):710-5; discussion 715-6.
- C R Boland, S N Thibodeau, S R Hamilton, D Sidransky, J R Eshleman, R W Burt, S J Meltzer, M A Rodriguez-Bigas, R Fodde, G N Ranzani, S Srivastava. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998 Nov 15;58(22):5248-57.
- Stoffel E M, Mangu P , Gruber S , Hamilton S , Kalady M , Wan Yee Lau M , Lu K, Roac N, American Society of Clinical Oncology; European Society of Clinical Oncology. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol 2015 Jan 10;33(2):209-17



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}