a cura di Odile Correnti – Medico Oftalmologo
Dal nome di un oculista scozzese Charles Usher, la Sindrome di Usher, considerata una malattia rara congenita, è stata studiata in realtà per la prima volta dal medico Von Graefe nel 1858.
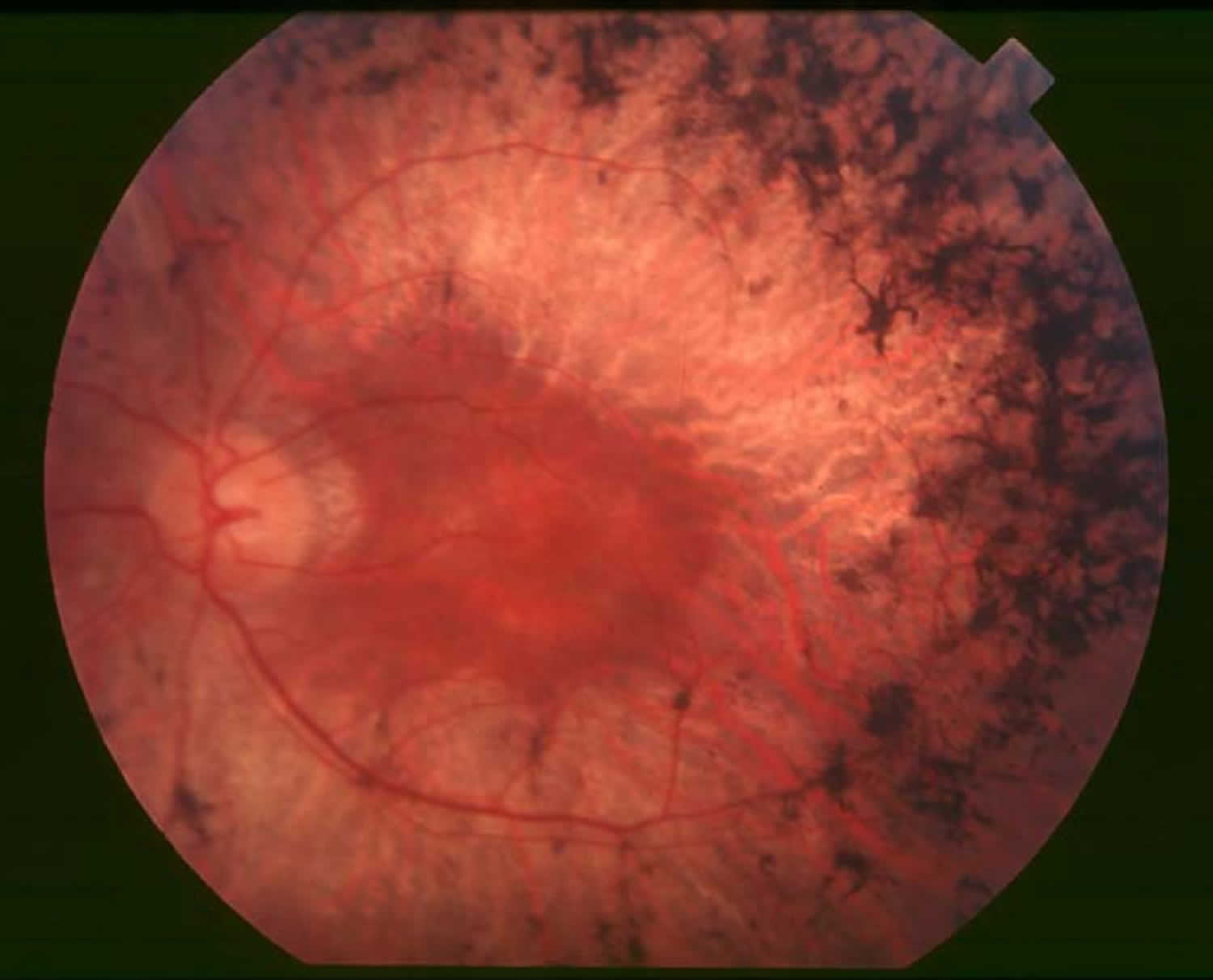
La Sindrome di Usher si manifesta con sordità neurosensoriale (di solito ipoacusia congenita) associata a retinite pigmentosa. Si stima che la sua prevalenza sia 1/30.000.
Si tratta della causa più comune di cecità associata a sordità a esordio nell’infanzia.
In base alle caratteristiche cliniche, all’ipoacusia, alla retinite pigmentosa e in alcuni casi ai deficit vestibolari (e dunque problemi di equilibrio), se ne riconoscono tre tipi.
Forme cliniche
I pazienti affetti da Sindrome di Usher Tipo I (circa il 40% dei casi) nascono con una sordità congenita profonda, non progressiva.
Possono talora conservare un residuo uditivo per le basse frequenze, che però non permette loro di udire; si associa areflessia vestibolare per cui accusano disturbi dell’equilibrio in quanto non ricevono i segnali dall’orecchio interno. Verso la fine della prima decade di età, manifestano i primi sintomi della retinite pigmentosa, fotosensibilità e cecità crepuscolare.
Man mano il deficit del campo visivo si accentua sino ad una visione tubulare e in età adulta insorge più prematuramente la cataratta. I bambini che ne sono affetti posticipano le normali tappe dello sviluppo sia del linguaggio (generalmente usano la Lingua dei Segni) sia nel controllo del capo, nell’acquisizione della posizione seduta e della deambulazione autonoma.
I pazienti affetti da Sindrome di Usher Tipo II sono circa il 60% dei casi e nascono con una perdita uditiva bilaterale da moderata a grave. Tale sordità prelinguale ha una lenta progressione e non è associata ad alterazioni vestibolari. L’utilizzo dell’apparecchio acustico permette loro di sentire e sviluppare il linguaggio. La perdita di udito è stazionaria nella maggior parte dei casi; molto raramente si perde completamente la capacità uditiva. Il progredire della degenerazione retinica inizia dopo l’adolescenza.
La Sindrome di Usher tipo III è il tipo più raro ( < 3% dei casi), maggiormente conosciuto nei paesi scandinavi e infrequente al di fuori di queste nazioni. Chi ne risulta affetto nasce senza manifestare un deficit uditivo, ma l’ipoacusia, di solito diagnosticata nella prima decade di vita, è rapidamente progressiva e intorno ai 30-50 anni può diventare profonda. Si associa nella metà dei casi disfunzione vestibolare.
La retinite pigmentosa, che in genere viene diagnosticata dopo la sordità, nella fase iniziale comporta deficit visivo in presenza di luce scarsa e tende a degenerare verso la completa cecità nell’arco di qualche decennio.
La diagnosi clinica, soprattutto per i primi 2 tipi, si basa sulle caratteristiche e la tipologia di sordità neurosensoriale bilaterale
- simmetrica, congenita e profonda per il tipo 1
- moderata e grave con una importante perdita neurosensoriale alle alte frequenze per il tipo 2
Base genetica
In tutti e 3 i casi la trasmissione della Sindrome di Usher è autosomica recessiva.
Le ricerche genetiche effettuate negli ultimi dieci anni, hanno dimostrato che i tre tipi di Sindrome di Usher si dividono in diversi sottotipi (vedi tabella ). Attualmente è stato verificato che a giocare un ruolo determinante nello sviluppo della Sindrome di Usher sono undici loci (cromosomi) e otto geni.
Il sottotipo USH1b, uno dei più comuni, presenta la mutazione del gene myosin VIIA, che si esprime a livello dei fotorecettori retinici e della coclea interessando le cellule ciliate; anche il gene uscerin (sottotipo USH2a) interessa le stesse aree sebbene a livello della coclea coinvolga le cellule connettive.
| TIPO | SOTTOTIPO GENETICO | GENE | CROMOSOMA |
| Tipo I Usher | USH1a | — | 14 |
| USH1b | MYO7A | 11 | |
| USH1C | USH1C | 11 | |
| USH1d | CDH23 | 10 | |
| USH1e | — | 21 | |
| USH1f | PCDH15 | 10 | |
| USH1g | SANS | 17 | |
| Tipo II Usher | USH2a | USH2A | 1 |
| USH2b | — | 3 | |
| USH2c | VLGR1 | 5 | |
| Tipo III Usher | USH3 | CLRN1 | 3 |
Come si evince più schematicamente dalla tabella, il sottotipo clinico 1 è dovuto alle mutazioni di cinque geni (MYO7A, USH1C, CDH23, PCDH15, USH1G) e di un locus (USH1E); il sottotipo 2 è dovuto alle mutazioni di tre geni (USH2A, GPR98 e DFNB31) e di un locus (15q); nel sottotipo 3 è mutato un solo gene (CLRN1).
L’esame genetico è possibile dopo avere effettuato indagini preliminari, alle quali segue la diagnosi molecolare basata sulla sequenza genomica dei geni-malattia.
La consulenza genetica è estremamente importante: le mutazioni eterozigoti del gene USH2A sono relativamente frequenti e dunque è giusto renderne edotti i genitori. La diagnosi prenatale è possibile nelle famiglie nelle quali sia stata identificata la mutazione patogenetica mediante diagnosi molecolare basata sulla sequenza genomica dei geni-malattia. La diagnosi differenziale si pone con le sindromi oculo-acustiche associate a mutazioni mitocondriali del DNA (MIDD, sindrome di Kearns-Sayre) e, più raramente, con la malattia di Refsum o con forme attenuate della sindrome di Alström.
L’approccio multidisciplinare
È indubbio che i pazienti affetti da Sindrome di Usher debbano necessariamente essere approcciati su base multidisciplinare, per la gestione sia della sordità che della cecità che dei disturbi del linguaggio (otorinolaringoiatri, oculisti, logopedisti, psicologi, specialisti delle protesi acustiche).
Sono necessari programmi di sviluppo psicomotorio e di apprendimento personalizzati per i pazienti sordo-ciechi. Gli impianti cocleari, monolaterali e bilaterali, sono ampiamente usati per i pazienti con sordità profonda; gli impianti cocleari e le protesi acustiche risultano più efficaci se impiantati in tenera età.
Gli ausilii per ipovisione nelle fasi avanzate della malattia possono essere di valido aiuto. Sono in corso ricerche in termini di terapia genica, neuroprotezione e sistemi visivi artificiali.
La prognosi della malattia dipende dalla progressione del deficit visivo: la cecità si verifica in quasi tutti i casi tra i 50 e i 70 anni di vita.
Una diagnosi tempestiva della sindrome di Usher può ridurne sul paziente l’effetto psicologico: con il sostegno e la professionalità di un team esperto e soprattutto con il valido aiuto della famiglia, è fondamentale che il paziente acquisti e/o mantenga la propria indipendenza, e nonostante le insormontabili difficoltà, viva la sua vita con grande entusiasmo e coraggio.
BIBLIOGRAFIA
- Eandi CM, Dallorto L, Spinetta R, Micieli MP, Vanzetti M, Mariottini A, Passerini I, Torricelli F, Alovisi C, Marchese C. Targeted next generation sequencing in Italian patients with Uscher syndrome: phenotype-genotype correlations. Sci Rep. 2017 Nov 15;7(1):15681.
- Hossain MM, Islam MF. Usher syndrome. Mymensingh Med J. 2012 Jan;21(1):155-7.
- Seeliger MW, Fischer MD, Pfister M. Usher Syndrome: clinical features, diagnostic options, and therapeutic prospects. Ophthalmologe. 2009 Jun; 106 (6): 505-11.
- Tazetdinov AM, Dzhemileva LU, Khusnutdinova EK. Molecular genetics of Usher Syndrome. Genetika 2008. Jun; 44 (6): 725-33.
- Geng R, Omar A, Gopal SR, Chen DH, Stepanyan R, Basch ML, Dinculescu A, Furness DN, Saperstein D, Hauswirth W, Lustig LR, Alagramam KN. Progressive Modeling and Preventing Hearing Loss in Usher Syndrome IIISee comment in PubMed Commons below. Sci Rep. 2017 Oct 18;7(1):13480. 13620-9.
- Magliulo G, Iannella G, Gagliardi S, Iozzo N, Plateroti R, Mariottini A, Torricelli F. Usher’s Syndrome Type II: A Comparative Study of Genetic Mutations and Vestibular System Evaluation. Otolaryngol Head Neck Surg. 2017 Nov;157(5):853-860.
- Vozzi D, Aaspõllu A, Athanasakis E, Berto A, Fabretto A, Licastro D, Külm M, Testa F, Trevisi P, Vahter M, Ziviello C, Martini A, Simonelli F, Banfi S, Gasparini P. Molecular epidemiology of Usher syndrome in Italy. Mol Vis. 2011;17:1662-8.
- Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher syndrome: hearing loss with vision loss. Adv Otorhinolaryngol. 2011;70:56-6



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}