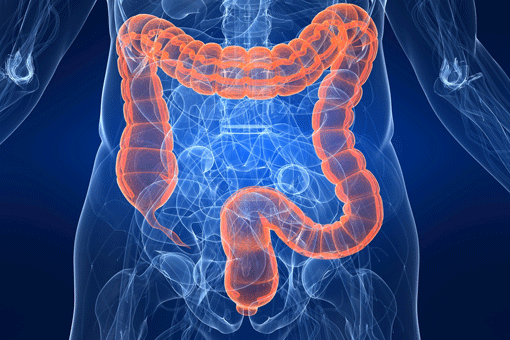
Caratteristiche e sintomi
La poliposi adenomatosa familiare del colon (FAP) è caratterizzata dalla comparsa, generalmente
sin dalla seconda decade di vita, di centinaia o migliaia di adenomi nel retto e nel colon. In molti
pazienti la condizione rimane asintomatica per diversi anni sino a quando, per l’aumentare del numero e
della dimensione degli adenomi, compare generalmente rettoragia e anemia. Alcuni sintomi aspecifici che possono far sospettare la presenza della poliposi, sono rappresentati da costipazione o diarrea,
dolori addominali, masse palpabili all’addome e calo ponderale.
La FAP può comportare anche dei sintomi extraintestinali come osteomi, anomalie dei denti, ipertrofia congenita dell’epitelio pigmentato della retina, tumori desmoidi e tumori non localizzati nel colon (tiroide, fegato, dotti biliari, sistema nervoso centrale). Una variante meno aggressiva è la FAP attenuata (AFAP) caratterizzata da un numero inferiore di polipi adenomatosi colon-rettali (di solito tra 10 e 100), con comparsa di adenomi in età più tardiva e basso rischio di cancro. La poliposi adenomatosa familiare interessa 1 persona su 8.000-14.000. I polipi sono presenti nel 50% dei soggetti entro i 15 anni d’età e nel 95% dei soggetti entro i 35 anni d’età.
Aspetti genetici
La forma classica di FAP si trasmette con modalità autosomica dominante ed è dovuta a mutazioni
germinali nel gene APC che è un gene oncosoppressore. Tale gene svolge un ruolo fondamentale nella via di segnalazione WNT regolando la fosforilazione della beta-catenina la quale deve essere successivamente degradata. Le mutazioni a livello di APC favoriscono la traslocazione di beta-catenina nel nucleo con la successiva trascrizione di alcuni proto-oncogeni, quali c-myc e ciclina D1, che hanno un ruolo centrale nel processo di cancerogenesi.
Esistono altri geni coinvolti nell’eziologia della poliposi del colon e tra questi il gene MUTYH che causa una poliposi autosomica recessiva, caratterizzata da un leggero aumento del rischio di sviluppare tumori del colon-retto e polipi/adenomi nel tratto gastrointestinale superiore e inferiore e geni quali SMAD4 e TP53 che causano forme più rare. La diagnosi differenziale si pone con altre malattie con polipi multipli (sindrome di Peutz-Jeghers e di Lynch, poliposi giovanile familiare o iperplastica, sindrome poliposica mista ereditaria).
Consulenza oncogenetica
In presenza di indicazione clinica o familiare è fondamentale eseguire una consulenza oncogenetica per ricostruire l’albero genealogico, valutare la storia familiare e personale e indirizzare l’analisi genetica. L’utilizzo di pannelli multigenici per la valutazione del rischio oncogenetico potrebbe permettere di ottimizzare la resa dell’indagine genetica. Individuata la mutazione genetica in un soggetto sarà possibile eseguire la ricerca single site della mutazione familiare negli altri soggetti.
Prevenzione e cura
Le persone affette da FAP hanno un rischio del 100% di sviluppare tumori del colon-retto. Tuttavia, tale rischio si riduce di molto quando i pazienti sono presi in carico e trattati precocemente. Si stima infatti che il rischio di trasformazione in carcinoma è molto basso in caso di adenomi inferiori ai 5 mm. Il rilievo di polipi > 1cm è associato ad un rischio neoplastico del 47% e polipi superiori a 2 cm di solito contengono già focolai di carcinoma. Oltre alla dimensione del polipo l’età del soggetto influenza il rischio di degenerazione neoplastica.
Sono state sviluppate Linee Guida specifiche come quelle dell’AIOM per la diagnosi precoce e la prevenzione. In presenza di mutazione genetica identificata i portatori della mutazione seguiranno uno specifico programma di sorveglianza che inizia già intorno agli 11-14 anni.
Da alcuni anni sono attivi diversi protocolli di ricerca che hanno ipotizzato anche l’utilizzo di alcune molecole per rallentare la proliferazione dei polipi e la loro degenerazione.
Per ulteriori approfondimenti puoi contattare i genetisti di Bgenetica o, qualora dovessi rientrare in una delle condizioni prima illustrate, valutare la possibilità di eseguire una consulenza genetica.
Se hai dei quesiti i Genetisti di Bgenetica sono pronti a risponderti inviando il tuo quesito a info@bgenetica.it
Bibliografia
- Sokic-Milutinovic A. Appropriate Management of Attenuated FamilialAdenomatous Polyposis: Report of a Case and Review of the Literature. Dig Dis. 2019;37(5):400-405.
- Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, Phillips NJ, Lai J, Guzman MA. FamilialAdenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. .Arch Pathol Lab Med. 2019 Nov;143(11):1382-1398
- Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, de Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. 2018 Jan;50(1):49-59
- Aihara H, Kumar N, Thompson CC. Diagnosis, surveillance, and treatment strategies for familialadenomatous polyposis: rationale and update. Eur J Gastroenterol Hepatol. 2014 Mar;26(3):255-62
- Sarvepalli S, Burke CA, Monachese M, Leach BH, Laguardia L, O’Malley M, Kalady MF, Church JM. Natural history of colonic polyposisin young patients with familial adenomatous polyposis. Gastrointest Endosc. 2018 Oct;88(4):726-733
- Hyer W, Cohen S, Attard T, Vila-Miravet V, Pienar C, Auth M, Septer S, Hawkins J, Durno C, Latchford A Management of FamilialAdenomatous Polyposis in Children and Adolescents: Position Paper From the ESPGHAN Polyposis Working Group. .J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):428-441.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}