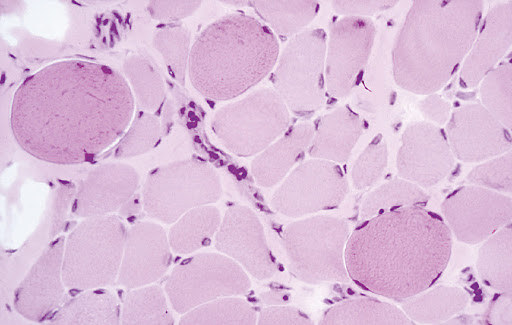
Caratteristiche
La Distrofia Muscolare di Duchenne (DMD) è una grave malattia genetica caratterizzata da atrofia muscolare progressiva, degenerazione dei muscoli scheletrici, lisci e cardiaci.
La DMD, vista la trasmissione X-linked, si manifesta prevalentemente nei maschi con un’incidenza di circa 1/3.300 nati maschi. Le femmine di solito sono asintomatiche, anche se una piccola parte delle portatrici presenta segni clinici lievi della malattia. L’esordio avviene nella prima infanzia e i bambini affetti possono presentare ritardo nelle tappe dello sviluppo motorio o ritardo globale.

Sintomi della distrofia muscolare
I primi sintomi della patologia si manifestano intorno ai tre anni: il bambino ha difficoltà nel correre, salire le scale, saltare, e mostra il cosiddetto “segno di Gowers”, un modo particolare di utilizzare le mani poggiate sulle cosce per alzarsi da terra o dalla posizione seduta. Nei pazienti che non eseguono alcun trattamento, la perdita della deambulazione autonoma avviene, generalmente, tra i 6 e i 13 anni. La diagnosi precoce e essenziale per salvaguardare la funzione muscolare il più a lungo possibile.
La diagnosi viene sospettata in base al quadro clinico, alla storia familiare e agli esami di laboratorio (la creatinchinasi sierica – CK – è 100-200 volte superiore ai livelli normali). Man mano che il danno muscolare progredisce, il tessuto connettivo e il grasso rimpiazzano le fibre muscolari, portando inesorabilmente all’indebolimento dei muscoli. I pazienti affetti da DMD normalmente perdono la capacità di camminare nella prima adolescenza, necessitano di supporto ventilatorio verso la fine dell’adolescenza e, infine, muoiono per insufficienza cardiaca e polmonare.

Aspetti genetici
La distrofia muscolare di Duchenne fu descritta per la prima volta nel 1868 dal neurologo francese Guillaume Duchenne de Boulogne. La base genetica venne invece identificata nel 1986. Il danno muscolare è dovuto all’assenza completa della distrofina, una proteina del sarcolemma, secondaria a mutazioni o delezioni del gene DMD (Xp21.2). Il rischio di ricorrenza è del 50% per le femmine portatrici di avere un figlio maschio affetto e del 50% di avere una figlia femmina portatrice.
Nelle famiglie in cui è stata identificata la presenza di soggetti portatori di alterazioni del gene DMD è possibile eseguire la diagnosi prenatale o valutare percorsi di PMA con diagnosi preimpianto. Tuttavia, circa un terzo dei casi di distrofia muscolare nasce da madri che non sono portatrici. In questo caso, la malattia è dovuta a una nuova mutazione o delezione del gene per la distrofina.
Al fine di individuare soggetti di sesso femminile portatrici di alterazioni del gene DMD all’interno di famiglie in cui non si sia mai manifestata la presenza di soggetti affetti, da recente, si stanno implementando strategie di analisi molecolari all’interno dei test genetici Carrier Screening.
La Distrofia Muscolare di Becker
Mutazioni del gene della distrofina possono comportare anche una forma più lieve clinicamente denominata Distrofia Muscolare di Becker (BMD). Ciò che differenzia la distrofia muscolare di Duchenne da quella di Becker è la quantità di distrofina funzionale prodotta nelle cellule muscolari: un’assenza completa della proteina determina la forma di Duchenne, mentre un’alterazione quantitativa o qualitativa di minore entità conduce alla distrofia di Becker. La BMD è una forma più lieve caratterizzata da un esordio tardivo e da un decorso meno definito. Un’ulteriore differenza tra la DMD e la BMD si riscontra nell’incidenza, la BMD ha un’incidenza nettamente minore, inferiore a 1 caso su 18.000, circa quattro volte meno della DMD.
Prospettive di cura
Ad oggi non esiste ancora una cura risolutiva per la malattia di Duchenne, ma la messa a punto di un approccio multidisciplinare, che comprende l’utilizzo di alcuni farmaci quali i corticosteroidei, la fisioterapia, la chirurgia ortopedica, la prevenzione cardiologia e l’assistenza respiratoria, ha permesso di limitare gli effetti della malattia e di migliorare le condizioni di vita. In un decennio, le aspettative di vita sono ormai raddoppiate ed iniziano a prospettarsi alcune terapie specifiche legate ad alcune forme genetiche come quelle legate alla presenza di mutazioni non senso che rappresentano circa il 10-15% dei casi. Per quanto riguarda la terapia vi sono numerose strategie che sono oggetto di diversi progetti di ricerca internazionali che mirano a fornire ai pazienti la distrofina o a correggere le mutazioni genetiche (come la terapia genica, la terapia cellulare, l’editing genomico, l’exon skipping o piccole molecole che permettono di mascherare le mutazioni).
Ad esempio, nel 2016, la FDA ha approvato, negli Stati Uniti, il trattamento con eteplirsen nei pazienti Duchenne che hanno mutazioni nel gene della distrofina trattabili con il salto dell’esone 51, mutazioni che colpiscono circa il 13% circa della popolazione DMD. Attualmente, l’unica molecola ad aver ricevuto l’approvazione in Europa è ataluren, una molecola che agisce esclusivamente sulle mutazioni “nonsenso”, che causano l’interruzione anticipata della lettura del gene e, quindi, la mancata produzione di distrofina funzionale. Le mutazioni “nonsenso” si trovano nel 10% circa della popolazione DMD.
L’analisi del DNA e l’identificazione della mutazione è quindi fondamentale perché permette di avere certezza della diagnosi, avviare un corretto follow up e accedere a potenziali terapie specifiche per alcune specifiche forme genetiche.
Per ulteriori approfondimenti puoi contattare i genetisti di Bgenetica o, qualora dovessi rientrare in una delle condizioni prima illustrate, valutare la possibilità di eseguire una consulenza genetica.
Se hai dei quesiti i Genetisti di Bgenetica sono pronti a risponderti inviando il tuo quesito a info@bgenetica.it
Bibliografia
- Salmaninejad A, Valilou SF, Bayat H, Ebadi N, Daraei A, Yousefi M, Nesaei A, Mojarrad M Duchenne muscular dystrophy: an updated review of common available therapies. .Int J Neurosci. 2018 Sep;128(9):854-864
- Werneck LC, Lorenzoni PJ, Ducci RD, Fustes OH, Kay CSK, Scola RH. Duchenne muscular dystrophy: an historical treatment review. Arq Neuropsiquiatr. 2019 Sep 5;77(8):579-589.
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR; DMD Care Considerations Working Group.Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-267.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}