A cura di Claudia Di Napoli – Medico Genetista
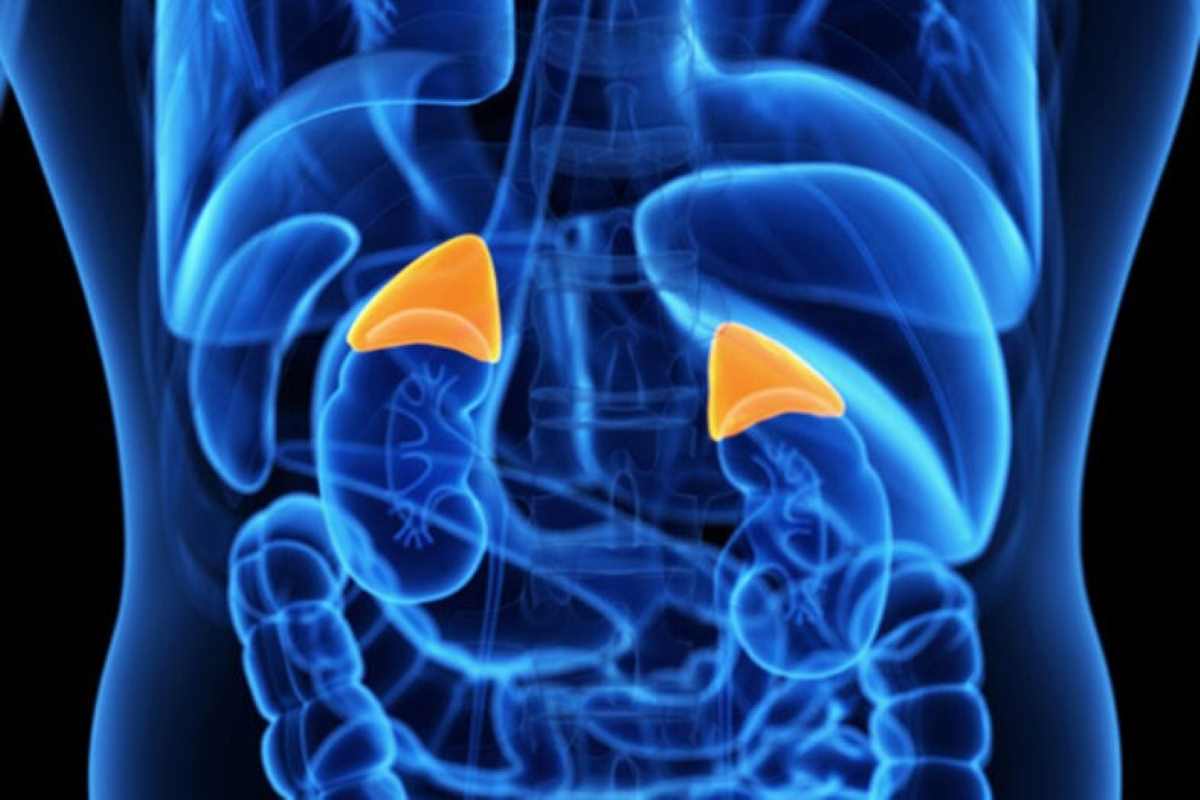
L’iperplasia surrenalica congenita (CAH) è una malattia endocrina ereditaria, con segni variabili di iper- o ipo-androgenismo, legati al tipo ed alla gravità, dovuta da un difetto nella produzione, da parte delle ghiandole surrenali (insufficienza surrenalica), di ormoni steroidei.
Si stima che la CAH si presenti con un’incidenza di circa 1 su 5.000-15.000 nati vivi.
Cause e modalità di trasmissione
La CAH è causata da varianti in alcuni geni che codificano per enzimi coinvolti nella sintesi degli ormoni steroidei.
Nel 90-95% dei casi, la CAH è causata da una variante nel gene CYP21A2, localizzato sul cromosoma 6p21.3. Il gene CYP21A2 codifica per un enzima che controlla il cortisolo e la produzione di aldosterone. Il deficit enzimatico che consegue alla presenza del difetto genetico comporta un accumulo dei precursori, che, soprattutto nel deficit dell’enzima 21-idrossilasi, vengono convogliati verso la sintesi di altri ormoni, ovvero quelli androgeni prodotti dalle ghiandole surrenali.
La CAH è una malattia autosomica recessiva, entrambe le copie del gene devono essere mutate e pertanto una coppia di genitori, portatori sani, ha un rischio del 25% per ogni gravidanza di concepire un figlio affetto. In presenza di familiarità è quindi indicata la consulenza genetica con l’esecuzione del test molecolare.
Sono state descritte varianti in altri geni, che sono responsabili di forme meno frequenti e che determinano le seguenti varianti: CAH da deficit di 17-alfa-idrossilasi, deficit di 3-beta-idrossisteroido deidrogenasi, deficit di 11-beta-idrossilasi, deficit di citocromo P450 ossidoreduttasi e iperplasia surrenalica lipoide congenita.
Clinica e Sintomi
Clinicamente è possibile distinguere più forme per la CAH da deficit di 21-idrossilasi:
- Forma classica:
- Perdita di sali: vomito, disidratazione, ipotensione e shock. Potenzialmente fatale.
- Virilizzazione: genitali ambigui e livelli variabili di virilizzazione nelle femmine. I genitali esterni nei maschi sono normali, si può osservare pubarca prematuro ed accelerazione della velocità di crescita e della maturazione scheletrica (con bassa statura in età adulta).
- Forma non classica:
- Sintomi più lievi: nelle femmine acne, irsutismo, irregolarità mestruali e/o anovulazione. I maschi (e alcune femmine) sono asintomatici.
Diagnosi dell’iperplasia surrenalica congenita
Nei soggetti di sesso femminile con CAH classica, il sospetto diagnostico è posto solitamente alla nascita per la presenza di genitali ambigui.
L’iter diagnostico prevede in ogni caso alla nascita:
- In molti paesi europei e in alcune regioni italiane (Veneto, Lombardia, Emilia Romagna, Valle d’Aosta e Piemonte) screening neonatale: misura dei livelli di 17-OH-progesterone.
- Dosaggio ormonale: cortisolo, ACTH.
- Test genetici: confermano la diagnosi.
Terapie
Con un trattamento adeguato, i pazienti possono avere un’aspettativa di vita normale.
Gli approcci terapeutici includono:
- Terapia sostitutiva con corticosteroidi (idrocortisone): per trattare l’insufficienza surrenalica, compensare il deficit e ridurre i livelli degli ormoni androgeni.
- Terapia sostitutiva con mineralocorticoidi (9 alfa-fludrocortisone acetato): in caso di forma classica con perdita di sali.
- Chirurgia: per correggere anomalie genitali nei casi necessari.
- Supporto psicologico.
Associazioni per Pazienti
- AISAC Onlus: https://airisc.org/home.html
Bibliografia
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018 Nov 1;103(11):4043-4088. doi: 10.1210/jc.2018-01865. Erratum in: J Clin Endocrinol Metab. 2019 Jan 1;104(1):39-40. doi: 10.1210/jc.2018-02371. PMID: 30272171; PMCID: PMC6456929.
- Nimkarn S, Gangishetti PK, Yau M, et al. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia. 2002 Feb 26 [Updated 2016 Feb 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1171/
- Husebye ES, Allolio B, Arlt W, Badenhoop K, Bensing S, Betterle C, Falorni A, Gan EH, Hulting AL, Kasperlik-Zaluska A, Kämpe O, Løvås K, Meyer G, Pearce SH. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med. 2014 Feb;275(2):104-15. doi: 10.1111/joim.12162. Epub 2013 Dec 16. PMID: 24330030.
- Orphanet: https://www.orpha.net/en/disease/detail/418



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}