A cura di Claudia Di Napoli – Medico Genetista
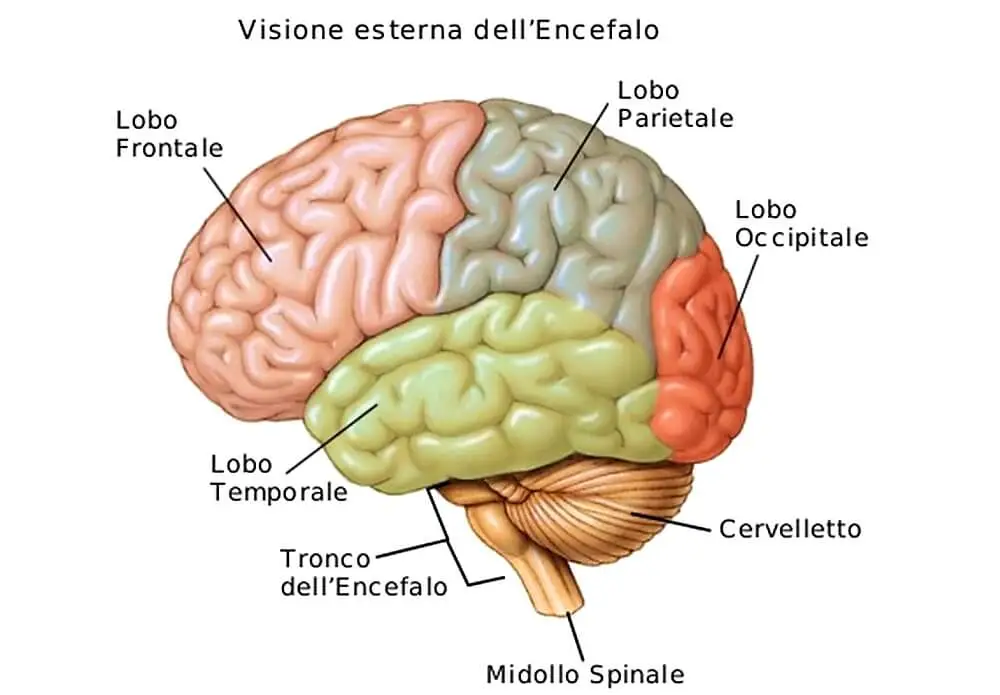
Le atassie spinocerebellari (SCA) rappresentano un gruppo eterogeneo di malattie neurodegenerative ereditarie che colpiscono il cervelletto, il midollo spinale e talvolta altre aree del sistema nervoso centrale. Queste patologie sono caratterizzate da una progressiva perdita di coordinazione motoria (atassia) che può coinvolgere il cammino, la parola e i movimenti oculari. Esistono oltre 40 sottotipi di SCA, classificati principalmente in base alle mutazioni genetiche responsabili (es. SCA1, SCA2, SCA3, etc.).
Epidemiologia SCA
Le SCA sono considerate malattie rare, con una prevalenza stimata tra 1 e 5 casi ogni 100.000 persone, anche se la frequenza può variare significativamente a seconda della popolazione studiata. In Italia le forme più frequenti sono la SCA1 e la SCA2.
Cause SCA
Le SCA sono più frequentemente causate da mutazioni genetiche trasmesse con modalità autosomica dominante, il che significa che è sufficiente una copia del gene mutato per sviluppare la malattia e che un soggetto affetto ha il 50% di probabilità di trasmettere la malattia alla prole. Molti sottotipi sono associati a un fenomeno chiamato “espansione di triplette”, in cui una sequenza di nucleotidi (CAG) nel DNA è ripetuta più volte del normale, portando alla produzione di proteine anomale che danneggiano le cellule nervose. Le patologie da ripetizione di triplette sono inoltre caratterizzate dal fenomeno dell’anticipazione, ovvero l’aumento del numero di triplette nella trasmissione da una generazione all’altra con insorgenza dei sintomi in età più precoce.
Sono inoltre stati descritti anche casi meno frequenti di trasmissione autosomica recessiva, come per esempio la SCAR4, e X-linked.
Clinica SCA
Le manifestazioni cliniche delle SCA variano a seconda del sottotipo, ma i sintomi più comuni includono:
- Atassia del cammino: difficoltà a mantenere l’equilibrio e coordinare i movimenti.
- Disartria: difficoltà nel parlare chiaramente.
- Movimenti oculari anomali: nistagmo o difficoltà a seguire oggetti con gli occhi.
- Altri sintomi neurologici: tremore, rigidità, neuropatia periferica e, in alcuni casi, deterioramento cognitivo.
L’età di esordio della sintomatologia varia in base forma specifica di SCA; tuttavia esiste un’ampia variabilità dei segni clinici nei soggetti affetti, anche all’interno dello stesso nucleo familiare. La progressione è lenta ma inevitabile, con una crescente disabilità nel tempo.
Diagnosi SCA
La diagnosi di SCA si basa su:
- Esame clinico e neurologico: valutazione dei sintomi e della storia familiare.
- Esami strumentali: la risonanza magnetica (RM) mostra atrofia del cervelletto e delle altre strutture colpite.
- Test genetici: identificazione delle mutazioni responsabili, che confermano la diagnosi e determinano il sottotipo specifico.
Terapie SCA
Ad oggi, non esiste una cura per le SCA. Il trattamento è sintomatico e mira a migliorare la qualità di vita dei pazienti:
- Fisioterapia e terapia occupazionale: per mantenere la mobilità e l’autonomia.
- Logopedia: per migliorare la comunicazione e la deglutizione.
- Farmaci sintomatici: per gestire sintomi specifici come tremore o spasticità.
La ricerca è attiva nello sviluppo di terapie genetiche e farmacologiche mirate a rallentare la progressione della malattia.
Associazioni per Pazienti
In Italia, esistono diverse associazioni che supportano i pazienti affetti da SCA e le loro famiglie. Tra queste:
- AISA (Associazione Italiana per la lotta alle Sindromi Atassiche): offre supporto e informazione a livello nazionale. www.atassia.it
- UNIAMO (Federazione Italiana Malattie Rare): un network di associazioni per malattie rare. www.uniamo.org
Bibliografia
- Sullivan R, Yau WY, O’Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol. 2019 Feb;266(2):533-544. doi: 10.1007/s00415-018-9076-4. Epub 2018 Oct 3. PMID: 30284037; PMCID: PMC6373366. https://pubmed.ncbi.nlm.nih.gov/30284037/
- Ruano, L., Melo, C., Silva, M. C., & Coutinho, P. (2014). The global epidemiology of hereditary ataxias and spastic paraplegias: a systematic review of prevalence studies. Neuroepidemiology, 42(3), 174-183. https://pubmed.ncbi.nlm.nih.gov/24603320/
- PDTA RFG040 – Malattie Spinocerebellari. (2023). https://www.reteneuroscienze.it/wp-content/uploads/2024/05/Atassie-PDTA-RFG040_malattie-spinocerebellari-Pubblicato-29-Marzo-2023.pdf



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}